
.jpg "新闻中心")

反应操作、柱层析、蒸馏等实验室工作经验分享
发布时间:2023-05-19浏览次数:1551
实验室工作有很多是经验性的,有很多细节是课本上没有的,但是在实验室实际应用中又非常重要,本文我总结了一些自己在多年的实验室工作经验,主要是一些个人习惯。良好的实验室习惯的养成,往往会是一个很好的开始,它是避免人为反应失败的[敏感词]的利器,也是提高效率,避免人力浪费的很好的助力。
[敏感词]部分:反应操作
1,原则上所有的反应都不能用塞子塞住,从而造成反应在狭小的密闭空间下反应,必须在出口处街上氮气球或者三通(特殊的如高压反应或 sealed tube 反应除外),以免因反应放热或产生气体而发生冲料或者发生爆炸。
2,处理反应时选择合适的容器,[敏感词]在预估的所有体积的二到三倍之间,以免出现容器过满的状况。
3,任何反应若非会放出含 H+之物质,均应尽可能在惰气(氮气或氩气)系统下操作,以避免不必要之副反应(side reaction)。如必须于惰气系统下操作则以使用惰气气球系统为宜。
4,标准惰气系统反应处理方式是将反应器皿于烘箱中取出后迅速放入搅拌子并连接氮(氩)气出口之后迅速将反应所需设备组合起来,并用火焰干燥,然后抽真空再灌入氮(氩)气,并重复两次。
5,任何一未知反应,除非有极近似之反应作参考,否则应从0℃或室温开始尝试,若不反应再逐步升高其温度,反之若反应太快有副作用时,再降低其温度。
6,任何一未知反应于 set up 完成后30 分钟内即应检查反应进行之情况,我们可取适量之原液(Aliquots)以TLC,NMR,GC,IR 或其他适当的技巧检验之。确定其反应进度后,才可决定是否在原条件下反应,切不可任意由其反应,或work-up 而不予检查。
7,检查反应时若发现超过 3 小时没有任何变化(反应)发生则可尝试逐渐升高温度10-30℃,然后再等十分钟后检查其变化,若再过一小时后仍无反应则再升高温度10-30℃,如此反复尝试直到反应发生为止。
8,任何一反应若需加热或反应中可能生热时必须加装冷凝管以确保物质不会挥发散失。
9,若反应之溶剂为低沸点物质(如乙醚,二氯甲烷等)则冷凝管内需通冰水,否则溶剂可能挥发散失,尤其在加热回流时,不注意会造成反应物浓度过高,甚至干涸。
10,怕水而需低温反应,先Set-up 好所有的equipment、solvent,及部分不会产生反应之试剂,最后才以冷媒冷却反应系统,否则在set-up 时易有水汽进入,影响反应效果。
11,如果反应之产物 NMR 无法解释,应立即点TLC 以检查其纯度,如果不纯则NMR图谱之积分自然无法解释,应纯化后再测。
12,任何新反应在完全定案前,所有尝试反应之产物,请务必予以保留,即使非我们所预期的产物或混合物,均有保留及比较TLC 的价值。
13,反应后务必将所有“有意义”之TLC plates 按原尺寸画于笔记薄上,以便以后比较用。
14,下班前应尽力完成第二天实验之准备工作,如明日需用之干燥玻璃器皿,设备,或找好需用的试剂及溶剂等。
15,如预期反应需超过 12 小时,宜尽量利用下午或晚上set up 反应。如预期反应需超过36 小时,宜尽量利用周5 或周6 set up,以求节省时间提高效率。
第二部分:柱层析
1,一般而言极性大的物质在极性固定相上的移动速率最小(因其吸引力[敏感词])反之极性小的物质在极性固定相上的移动速率[敏感词]。但所有物质使用高极性流动相时在极性静相时之移动速率均增大。故依各分离物质的极性选择极性适当的固定相及流动相甚至其用量均为层析(Chromatography)成功之关键。

2,任何化学物若以 TLC 鉴定其纯度,其所显示的Rf 值超过0.7 或低于0.1 时,均无法确定其是否为单一点(one spot),因为在此种Rf 值时,许多Rf 较接近的点会重合。因此我们所选择观察物最适合之Rf 值为0.3-0.5。
3,Run Column 时所选用之solvent 系统应配合选用硅胶(stationary phase)的用量及sample(样品)的量来选择其极性组合,一般而言欲分离之主样品Rf 值以0.15-0.25为宜。
4,一般最常用之 Solvent 依其极性大小依次为n-Hexane(or pet.ether)<EtOAc<MeOH,组合为最通用,其他如Ether 也常为中极性之溶剂,对于氨类氯化溶剂如CH2Cl2,CHCl3常有特殊的效果,而Benzene 对含芳香性之化合物也会有出人意外之好效果。
5,Run Column 所用之Solvent 在配好后,应以TLC 再作检测一次,以确定是否为恰当之Solvent 系统。尤其若需要收集一点时,则在分离前之混合物必须留一些以便作比较参考用。
6,在填装层析管(packing column)时若不慎有气泡(如Solvent 部分流干)可补满Solvent后以木棒(或签字笔)轻敲管壁,让气泡浮出。因为气泡中没有硅胶,分离物流经此处时移动速度会比其他处快,而造成此区域分离物与其他无气泡处之分离物排列次序混乱之现象,影响分离效果。
7,在 Run Column 时所收集的分离份(fraction)体积(毫升)通常为硅胶克数的1/4 以下。实际情况可用下列公式来求出约略值,如欲分离成分A 与B,而A 之Rf 值为0.3,B 之Rf 值为0.2 则最适合之分离份(fraction)体积为:设硅胶量为50g
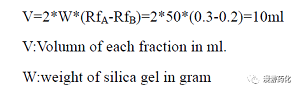
8,在作层析时,如何浓密而又均匀的将样品 Loading 在起始点乃是层析分离成功与否的关键。通常的做法是将样品溶解在不超过其体积两倍之低极性溶液,然后小心的将样品涂布于起始点。柱层析(Column Chromatography)时务必等样品液完全进入固定相(Stationary phase)后,以少许溶剂再将管壁上沾着的样品冲入固定相后,才可正式加溶剂开始 Run Column。由于大部分化合物在低极性溶剂中溶解度很差,我们也可以将样品均匀地与少量硅胶混合后干法上样。这也是我们采用最多的方法。
9,在 Run Column 时切记不可使Column 内溶剂过少而使静相顶端干涸,且在添加溶剂也要小心,不可使溶剂急冲而下破坏静相之顶层,尤其在刚开始Run Column 时,否则将造成样品之过量稀释分散,从而严重影响层析效果。
10,在收集时,可以先行以 Rf 值以固定相之总量计算大约样品出现时之流出溶剂体积,在此体积达1/2 量前可以大瓶收集,1/2 量之后再以预定之小容器分别收集。
11,Run Column 时Solvent 之流速一般而言(HPLC,MPLC 除外),流速均以越快越好,因为一般而言Mobile Phase Velocity 与Plate height 成反比,而Plate heigh 越小越好,故动相流速是越快越好。故Flash Chromatography 之效果常较一般Gravity column 为佳。
第三部分:蒸馏
1,蒸馏时务必先判断你们样品在其沸点时是否会分解,否则就必须选择减压蒸馏或其他的分离方法。
2,一般实验室的蒸馏仅用于分离沸点差 50℃以上物质。(通常只是用于除去溶剂或有色的高分子粘滞物)。如欲分离沸点较接近之物质则需加装分馏管,而分馏管之长度即装填物需视分离之物质沸点接近之程度而定。一般而言,沸点差小于20℃或总量小于1 克,则需特殊装置(如Spinning band or molecular distillation)否则便无法做分馏了。
3,蒸馏时其外加热媒之温度与蒸出沸点差至少会在 30℃左右。
4,在做减压蒸馏前,务请查明所有欲蒸馏物的沸点,以免于压力速降时造成‘突沸’而造成整个样品冲出。尤其需注意溶剂是否已在Rotary evaporator 上抽干。
5,如果无参考图表,可约略以下法计算各物质之低压沸点。通常压力由760mmHg 减至25mmHg 其沸点降低100℃,其后压力每降低一半,其沸点降低10℃。
6,若两个或更多物质混合时,通常会形成各种成分之共沸物。而在较低的温度即沸腾汽化而出。此种情形最常发生于样品含溶剂的状况。
7,减压蒸馏时,务必加装冷却 Trap,否则不但可能因样品被吸入泵而失去样品,也会损坏真空泵。
第四部分:其他日常操作
1,使用样品后尽可能将其归还回原位,尤其需冷藏或干燥者必须封好后放回原位。所有装于烧瓶内之化合物,应随时标明编号(以胶带粘贴以便清除)。
2,烘箱非储藏柜仅仅用于烘干玻璃器皿,烘干后需在近期内(一天内)使用者才放入,其余物品均应放于抽屉或凉干架。
3,如果 Solvent 或低沸点物(bp<150℃)能在Rotary evaporator 上清除者,必须先在Rotaryevaporator 上处理至不能再浓缩后,才可放上Vacuum pump。否则低沸点物可能被抽入泵中。
4,使用 Vacuum pump 必须要干冰作Trap 之冷媒,使用前必检查Trap 内是否有残留物,若有,则需清除后方可使用。并在使用完后立即清理Trap 之废物。
5,萃取时选用之 Solvent 应注意其比重,若其比重大于1(如CH2Cl2, CHCl3, CCl4)而反应原液为比重小于1 之溶剂,(如Ether, THF 或Benzene)时,切不可同时混入分液漏斗,否则会有乳化不分层之现象,造成分离困难。
6,当溶液装于有磨口的容器内时,在任何操作状况时,均应避免使溶液接触到磨口,因为当溶剂挥发后溶质将残留在磨口上,会使你损失所要的Compound,而在加盖常会有打不开之现象。所以若不慎或有不得以有溶液接触磨口,则应以纯溶剂浸润接触部分,务使溶剂残留之痕迹消除为止。
7,实验记录本应包含反应方程式,参考文献,试药用量(Weight,Mole),理论产率,实验步骤,TLC,光谱记录,与注意事项或结论。
8,实验室对量的记载应尽量保持三为数字,以求其[敏感词]性;对不满 1 之单位应用下一级之单位。如0.1 克应写为100mg,0.1mol 应写为100mmol。
9,实验本务必按进度如实记录,要如实做到‘做到哪里,写到哪里’,而且不得带出实验室。实验记录务必跟随实验进度将所有事实详细记录,成功之反应必有产物,产率,颜色,状态及纯化方式,失败之实验必说明鉴定失败的方法及证据,并尽可能说明原因。
上一篇:实验室常用干燥剂有哪些?